- Syllabus
- 1 Introduction
- 2 Data in Biology
- 3 Preliminaries
- 4 R Programming
- 4.1 Before you begin
- 4.2 Introduction
- 4.3 R Syntax Basics
- 4.4 Basic Types of Values
- 4.5 Data Structures
- 4.6 Logical Tests and Comparators
- 4.7 Functions
- 4.8 Iteration
- 4.9 Installing Packages
- 4.10 Saving and Loading R Data
- 4.11 Troubleshooting and Debugging
- 4.12 Coding Style and Conventions
- 4.12.1 Is my code correct?
- 4.12.2 Does my code follow the DRY principle?
- 4.12.3 Did I choose concise but descriptive variable and function names?
- 4.12.4 Did I use indentation and naming conventions consistently throughout my code?
- 4.12.5 Did I write comments, especially when what the code does is not obvious?
- 4.12.6 How easy would it be for someone else to understand my code?
- 4.12.7 Is my code easy to maintain/change?
- 4.12.8 The
stylerpackage
- 5 Data Wrangling
- 6 Data Science
- 7 Data Visualization
- 8 Biology & Bioinformatics
- 8.1 R in Biology
- 8.2 Biological Data Overview
- 8.3 Bioconductor
- 8.4 Microarrays
- 8.5 High Throughput Sequencing
- 8.6 Gene Identifiers
- 8.7 Gene Expression
- 8.7.1 Gene Expression Data in Bioconductor
- 8.7.2 Differential Expression Analysis
- 8.7.3 Microarray Gene Expression Data
- 8.7.4 Differential Expression: Microarrays (limma)
- 8.7.5 RNASeq
- 8.7.6 RNASeq Gene Expression Data
- 8.7.7 Filtering Counts
- 8.7.8 Count Distributions
- 8.7.9 Differential Expression: RNASeq
- 8.8 Gene Set Enrichment Analysis
- 8.9 Biological Networks .
- 9 EngineeRing
- 10 RShiny
- 11 Communicating with R
- 12 Contribution Guide
- Assignments
- Assignment Format
- Starting an Assignment
- Assignment 1
- Assignment 2
- Assignment 3
- Problem Statement
- Learning Objectives
- Skill List
- Background on Microarrays
- Background on Principal Component Analysis
- Marisa et al. Gene Expression Classification of Colon Cancer into Molecular Subtypes: Characterization, Validation, and Prognostic Value. PLoS Medicine, May 2013. PMID: 23700391
- Scaling data using R
scale() - Proportion of variance explained
- Plotting and visualization of PCA
- Hierarchical Clustering and Heatmaps
- References
- Assignment 4
- Assignment 5
- Problem Statement
- Learning Objectives
- Skill List
- DESeq2 Background
- Generating a counts matrix
- Prefiltering Counts matrix
- Median-of-ratios normalization
- DESeq2 preparation
- O’Meara et al. Transcriptional Reversion of Cardiac Myocyte Fate During Mammalian Cardiac Regeneration. Circ Res. Feb 2015. PMID: 25477501l
- 1. Reading and subsetting the data from verse_counts.tsv and sample_metadata.csv
- 2. Running DESeq2
- 3. Annotating results to construct a labeled volcano plot
- 4. Diagnostic plot of the raw p-values for all genes
- 5. Plotting the LogFoldChanges for differentially expressed genes
- The choice of FDR cutoff depends on cost
- 6. Plotting the normalized counts of differentially expressed genes
- 7. Volcano Plot to visualize differential expression results
- 8. Running fgsea vignette
- 9. Plotting the top ten positive NES and top ten negative NES pathways
- References
- Assignment 6
- Assignment 7
- Appendix
- A Class Outlines
8.3 Bioconductor
Bioconductor is an organized collection of strictly biological analysis methods packages for R. These packages are hosted and maintained outside of CRAN because the maintainers enforce a more rigorous set of coding quality, testing, and documentation standards than is required by normal R packages. These stricter requirements arose from a recognition that software packages are generally only as usable as their documentation allows them to be, and also that many if not most of the users of these packages are not statisticians or experienced computer programmers. Rather, they are people like us: biological analysis practitioners who may or may not have substantial coding experience but must analyze our data nonetheless. The excellent documentation and community support of the bioconductor ecosystem is a primary reason why R is such a popular language in biological analysis.
Bioconductor is divided into roughly two sets of packages: core maintainer packages and user contributed packages. The core maintainer packages are among the most critical, because they define a set of common objects and classes (e.g. the ExpressionSet class in the Biobase package) that all Bioconductor packages know how to work with. This common base provides consistency among all Bioconductor packages thereby streamlining the learning process. User contributed and maintained packages provide specialized functionality for specific types of analysis.
Bioconductor itself must be installed prior to installing other Bioconductor packages. To [install bioconductor], you can run the following:
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install(version = "3.14")Bioconductor undergoes regular releases indicated by a version, e.g. version = "3.14" at the time of writing. Every bioconductor package also has a version,
and each of those versions may or may not be compatible with a specific version
of Bioconductor. To make matters worse, Bioconductor versions are only
compatible with certain versions of R. Bioconductor version 3.14 requires R
version 4.1.0, and will not work with older R versions. These versions can cause
major version compatibility issues when you are forced to use older versions of
R, as may sometimes be the case on managed compute clusters. There is not a good
general solution for ensuring all your packages work together, but the general
best rule of thumb is to use the most current version of R and all packages at
the time when you write your code.
The BiocManager package is the only Bioconductor package installable using
install.packages(). After installing the BiocManager package, you may then
install bioconductor packages:
# installs the affy bioconductor package for microarray analysis
BiocManager::install("affy")One key aspect of Bioconductor packages is consistent and helpful documentation; every package page on the Bioconductor.org site lists a block of code that will install the package, e.g. for the affy package. In addition, Biconductor provides three types of documentation:
- Workflow tutorials on how to perform specific analysis use cases
- Package vignettes for every package, which provide a worked example of how to use the package that can be adapted by the user to their specific case
- Detailed, consistently formatted reference documentation that gives precise information on functionality and use of each package
The thorough and useful documentation of packages in Bioconductor is one of the reasons why the package ecosystem, and R, is so widely used in biology and bioinformatics.
The base Bioconductor packages define convenient data structures for storing and analyzing many types of data. Recall earlier in the Types of Biological Data section, we described five types of biological data: primary, processed, analysis, metadata, and annotation data. Bioconductor provides a convenient class for storing many of these different data types together in one place, specifically the SummarizedExperiment class in the package of the same name package. An illustration of what a SummarizedExperiment object stores is below, from the Bioconductor maintainer’s paper in Nature:
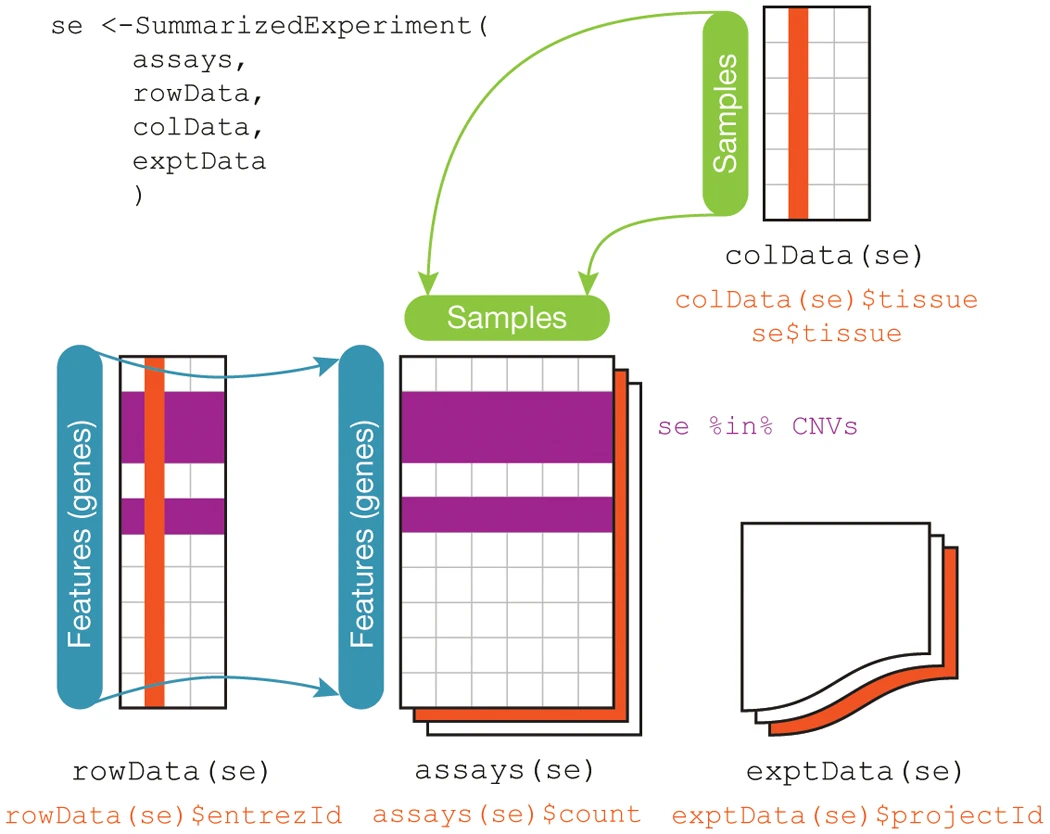
SummarizedExperiment Schematic - Huber, et al. 2015. “Orchestrating High-Throughput Genomic Analysis with Bioconductor.” Nature Methods 12 (2): 115–21.
As you can see from the figure, this class stores processed data (assays),
metadata (colData and exptData), and annotation data (rowData). The
SummarizedExperiment class is used ubiquitously throughout the Bioconductor
package ecosystem, as are other core classes some of which we will cover later
in this chapter.