- Syllabus
- 1 Introduction
- 2 Data in Biology
- 3 Preliminaries
- 4 R Programming
- 4.1 Before you begin
- 4.2 Introduction
- 4.3 R Syntax Basics
- 4.4 Basic Types of Values
- 4.5 Data Structures
- 4.6 Logical Tests and Comparators
- 4.7 Functions
- 4.8 Iteration
- 4.9 Installing Packages
- 4.10 Saving and Loading R Data
- 4.11 Troubleshooting and Debugging
- 4.12 Coding Style and Conventions
- 4.12.1 Is my code correct?
- 4.12.2 Does my code follow the DRY principle?
- 4.12.3 Did I choose concise but descriptive variable and function names?
- 4.12.4 Did I use indentation and naming conventions consistently throughout my code?
- 4.12.5 Did I write comments, especially when what the code does is not obvious?
- 4.12.6 How easy would it be for someone else to understand my code?
- 4.12.7 Is my code easy to maintain/change?
- 4.12.8 The
stylerpackage
- 5 Data Wrangling
- 6 Data Science
- 7 Data Visualization
- 8 Biology & Bioinformatics
- 8.1 R in Biology
- 8.2 Biological Data Overview
- 8.3 Bioconductor
- 8.4 Microarrays
- 8.5 High Throughput Sequencing
- 8.6 Gene Identifiers
- 8.7 Gene Expression
- 8.7.1 Gene Expression Data in Bioconductor
- 8.7.2 Differential Expression Analysis
- 8.7.3 Microarray Gene Expression Data
- 8.7.4 Differential Expression: Microarrays (limma)
- 8.7.5 RNASeq
- 8.7.6 RNASeq Gene Expression Data
- 8.7.7 Filtering Counts
- 8.7.8 Count Distributions
- 8.7.9 Differential Expression: RNASeq
- 8.8 Gene Set Enrichment Analysis
- 8.9 Biological Networks .
- 9 EngineeRing
- 10 RShiny
- 11 Communicating with R
- 12 Contribution Guide
- Assignments
- Assignment Format
- Starting an Assignment
- Assignment 1
- Assignment 2
- Assignment 3
- Problem Statement
- Learning Objectives
- Skill List
- Background on Microarrays
- Background on Principal Component Analysis
- Marisa et al. Gene Expression Classification of Colon Cancer into Molecular Subtypes: Characterization, Validation, and Prognostic Value. PLoS Medicine, May 2013. PMID: 23700391
- Scaling data using R
scale() - Proportion of variance explained
- Plotting and visualization of PCA
- Hierarchical Clustering and Heatmaps
- References
- Assignment 4
- Assignment 5
- Problem Statement
- Learning Objectives
- Skill List
- DESeq2 Background
- Generating a counts matrix
- Prefiltering Counts matrix
- Median-of-ratios normalization
- DESeq2 preparation
- O’Meara et al. Transcriptional Reversion of Cardiac Myocyte Fate During Mammalian Cardiac Regeneration. Circ Res. Feb 2015. PMID: 25477501l
- 1. Reading and subsetting the data from verse_counts.tsv and sample_metadata.csv
- 2. Running DESeq2
- 3. Annotating results to construct a labeled volcano plot
- 4. Diagnostic plot of the raw p-values for all genes
- 5. Plotting the LogFoldChanges for differentially expressed genes
- The choice of FDR cutoff depends on cost
- 6. Plotting the normalized counts of differentially expressed genes
- 7. Volcano Plot to visualize differential expression results
- 8. Running fgsea vignette
- 9. Plotting the top ten positive NES and top ten negative NES pathways
- References
- Assignment 6
- Assignment 7
- Appendix
- A Class Outlines
5.7 Arranging Data
After we have loaded our data from a file into a tibble, we often need to
manipulate it in various ways to make the values amenable to our desired
analysis. Such manipulations might include renaming poorly named columns,
filtering out certain records, deriving new columns using the values in others,
changing the order of rows etc. These operations may collectively be termed
arranging the data and many are provided in the
*dplyr package. We will cover some of the most
common data arranging functions here, but there are many more in the dplyr
package worth knowing.
In the examples below, we will make use of the following made-up tibble that contains fake statistics and p-values for three human genes:
gene_stats## # A tibble: 3 x 5
## gene test1_stat test1_p test2_stat test2_p
## <chr> <dbl> <dbl> <dbl> <dbl>
## 1 apoe 12.5 0.103 34.2 0.000013
## 2 hoxd1 4.40 0.632 16.3 0.0421
## 3 snca 45.7 0.0000000042 0.757 0.915The gene_stats tibble above is a simple example of a very common type of
data we work with in biology; namely instead of raw data, we work with
statistics that have been computed using raw data that help us interpret the
results. While these statistics may not be ‘data’ per se, we can still use all
the functions and strategies in the tidyverse to work with them.
The tidyverse is a very big place. RStudio created many helpful cheatsheets to aid in looking up how do to certain operations. The cheatsheet on dplyr has lots of useful information on how to use the many functions in the package.
5.7.1 dplyr::mutate() - Create new columns using other columns
Many biological analysis procedures perform some kind of statistical test on a
collection of features (e.g. genes) and produce p-values that indicate how
“surprising” each feature is according to the test. The p-values in our tibble
are nominal, i.e. they have not been adjusted for multiple
hypotheses. Briefly, when we run multiple tests like
we are on each of our three genes, there is a chance that some of the tests will
have a small p-value simply by chance. Multiple testing
correction
procedures adjust nominal p-values to account for this possibility in a number
of different ways, but the most common procedure in biological analysis is the
Benjamini-Hochberg or False Discovery Rate
(FDR) procedure. In R, the
p.adjust function can perform several of these procedures, including FDR:
dplyr::mutate(gene_stats,
test1_padj=p.adjust(test1_p,method="fdr")
)## # A tibble: 3 x 6
## gene test1_stat test1_p test2_stat test2_p test1_padj
## <chr> <dbl> <dbl> <dbl> <dbl> <dbl>
## 1 apoe 12.5 0.103 34.2 0.000013 0.155
## 2 hoxd1 4.40 0.632 16.3 0.0421 0.632
## 3 snca 45.7 0.0000000042 0.757 0.915 0.0000000126Notice how the adjusted p-values are larger than the nominal ones; this is the
effect of the multiple testing procedure. Since we have two sets of p-values,
we must compute the FDR on each of them, which we can do in the same call to
mutate():
gene_stats_mutated <- dplyr::mutate(gene_stats,
test1_padj=p.adjust(test1_p,method="fdr"),
test2_padj=p.adjust(test2_p,method="fdr")
)
gene_stats_mutated## # A tibble: 3 x 7
## gene test1_stat test1_p test2_stat test2_p test1_padj test2_padj
## <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
## 1 apoe 12.5 0.103 34.2 0.000013 0.155 0.000039
## 2 hoxd1 4.40 0.632 16.3 0.0421 0.632 0.0632
## 3 snca 45.7 0.0000000042 0.757 0.915 0.0000000126 0.915Another common operation is to create new columns derived from the values in
multiple other columns. We (or our wetlab colleagues) might decide it is
convenient to have a new column with TRUE or FALSE based on whether the
gene was significant in either test. Such a column would make it easy to filter
genes down to just ones that might be interesting in tools like Excel. We can
create new columns from multiple columns just as easily using the mutate()
function:
dplyr::mutate(gene_stats_mutated,
signif_either=(test1_padj < 0.05 | test2_padj < 0.05),
signif_both=(test1_padj < 0.05 & test2_padj < 0.05)
)## # A tibble: 3 x 9
## gene test1_stat test1_p test2_stat test2_p test1_padj test2_padj
## <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
## 1 apoe 12.5 0.103 34.2 0.000013 0.155 0.000039
## 2 hoxd1 4.40 0.632 16.3 0.0421 0.632 0.0632
## 3 snca 45.7 0.0000000042 0.757 0.915 0.0000000126 0.915
## # ... with 2 more variables: signif_either <lgl>, signif_both <lgl>Recall that the | and & operators execute ‘or’ and ‘and’ logic,
respectively. The above example required the creation of a new variable
gene_stats_mutated because the columns test1_padj and
test2_padj need to be in the tibble before computing the new fields. However,
in mutate(), columns created first in the function call are available to later
columns. In the following example, note that test1_padj is created first and
then used to create the signif columns:
dplyr::mutate(gene_stats,
test1_padj=p.adjust(test1_p,method="fdr"), # test1_padj created
test2_padj=p.adjust(test2_p,method="fdr"),
signif_either=(test1_padj < 0.05 | test2_padj < 0.05), #test1_padj used
signif_both=(test1_padj < 0.05 & test2_padj < 0.05)
)## # A tibble: 3 x 9
## gene test1_stat test1_p test2_stat test2_p test1_padj test2_padj
## <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
## 1 apoe 12.5 0.103 34.2 0.000013 0.155 0.000039
## 2 hoxd1 4.40 0.632 16.3 0.0421 0.632 0.0632
## 3 snca 45.7 0.0000000042 0.757 0.915 0.0000000126 0.915
## # ... with 2 more variables: signif_either <lgl>, signif_both <lgl>The alternative would be to split this into two mutate() commands, the first
creating the adjusted p-value columns and the second creating the significance
columns. dplyr recognizes how common it is to build new variables off of other
new variables in a mutate() command, and therefore provides this convenient
behavior.
mutate() can also be used to modify columns in place. The official convention
for human gene symbols is that they are upper case, but for some reason our
tibble contains lower case gene symbols. We can correct this using mutate()
but first we should talk about the stringr
package which makes working with strings much
easier than with base R functions.
5.7.2 stringr - Working with character values
Base R does not have very convenient functions for working with character strings (or just strings). This is due to its original intent a statistical programming language, where string manipulation is not (in principle) a common operation. However, in practice, we must frequently manipulate strings while loading, cleaning, and analyzing datasets. The stringr package aims to make working with strings “as easy as possible.”
The package includes many useful functions for operating on strings, including searching for patterns, mutating strings, lexicographical sorting, combining multiple strings together (i.e. concatenation), and performing complex search/replace operations. There are far too many useful functions to cover here and you should become comfortable reading the stringr documentation and the very helpful stringr cheatsheet.
In the previous section, we noted that the gene symbols in our tibble were lower
case while official gene symbols are in upper case. We can use the stringr
function stringr::str_to_upper() with the dplyr::mutate() function to
perform this adjustment easily:
dplyr::mutate(gene_stats,
gene=stringr::str_to_upper(gene)
)## # A tibble: 3 x 5
## gene test1_stat test1_p test2_stat test2_p
## <chr> <dbl> <dbl> <dbl> <dbl>
## 1 APOE 12.5 0.103 34.2 0.000013
## 2 HOXD1 4.40 0.632 16.3 0.0421
## 3 SNCA 45.7 0.0000000042 0.757 0.915Now are gene symbols have the appropriate case, and our wetlab colleagues won’t complain about it. :)
5.7.2.1 Regular expressions
Many of the string operations in the stringr package use regular
expression syntax. A regular
expression is a sequence of characters that describes patterns in text. Regular
expressions are written in a sort of mini programming language where certain
characters have special meaning that help in defining search patterns that
identifies the location of sequences of characters in text that follow a
particular pattern specified by the regular expression. This is similar to the
“Find” functionality in many word processors, but is more powerful due to the
flexibility of the patterns that can be found.
A simple example will be helpful. Let’s say we have a tibble containing the result of a (made-up) statistical test for all the genes in a genome:
de_genes## # A tibble: 39,535 x 4
## hgnc_symbol mean p padj
## <chr> <int> <dbl> <dbl>
## 1 MT-TF 5799 0.000910 0.00941
## 2 MT-RNR1 153 0.0272 0.0342
## 3 MT-TV 115 0.0228 0.0301
## 4 MT-RNR2 495 0.00318 0.0123
## 5 MT-TL1 20201 0.000377 0.00841
## 6 MT-ND1 160 0.0179 0.0258
## 7 MT-TI 3511 0.00247 0.0115
## 8 MT-TQ 772 0.00376 0.0129
## 9 MT-TM 301 0.00325 0.0124
## 10 MT-ND2 12 0.107 0.111
## # ... with 39,525 more rowsNow let’s say we’re interested in examining the results for the BRCA family of
genes, BRCA1 and BRCA2. We can use filter() on the data frame to look for them
individually:
de_genes %>% filter(hgnc_symbol == "BRCA1" | hgnc_symbol == "BRCA2")## # A tibble: 2 x 4
## hgnc_symbol mean p padj
## <chr> <int> <dbl> <dbl>
## 1 BRCA1 41 0.0321 0.0386
## 2 BRCA2 447 0.0140 0.0223This isn’t so bad, but we can do the same thing with
stringr::str_detect()
, which returns TRUE if the provided pattern matches the input and FALSE
otherwise, a regular expression, and the [dplyr::filter() function], which is
described in greater detail in a later section:
dplyr::filter(de_genes, str_detect(hgnc_symbol,"^BRCA[12]$"))## # A tibble: 2 x 4
## hgnc_symbol mean p padj
## <chr> <int> <dbl> <dbl>
## 1 BRCA1 41 0.0321 0.0386
## 2 BRCA2 447 0.0140 0.0223The argument "^BRCA[12]$" is a regular expression that searches for the
following:
- Search for genes that start with the characters
BRCA- the^at the beginning of the pattern stands for the start of the string - For genes that start with
BRCA, then look for genes where the next character is either1or2with[12]- the characters between the[]are searched for explicitly, and any character encountered that is not listed between them results in a non-match - For genes that start with
BRCAfollowed with either a1or a2, match successfully if the number is at the end of the name - the$at the end of the pattern stands for the end of the string
We can use these principles to find genes with more complex naming conventions. The Homeobox (HOX) genes encode DNA binding proteins that regulate gene expression of genes involved in morphgenesis and cell differentiation in vertebrates. In humans, HOX genes are organized into 4 clusters of paralogs that were the result of three DNA duplication events in the distant evolutionary past(Abbasi 2015), where each cluster encodes a subset of 13 distinct HOX genes placed next to each other. Each of these clusters has been assigned a letter identifier A-D (e.g. HOXA, HOXB, HOXC, and HOXD) and each paralogous gene within each cluster is assigned the same number (e.g. HOXA4, HOXB4, HOXC4, and HOXD4 are paralogs). There are 13 HOX genes in total, though not all genes remain in all clusters (e.g. HOXA1, HOXB1, and HOXD1 exist but HOXC1 was lost over time). The following figure depicts the human HOX gene clusters:
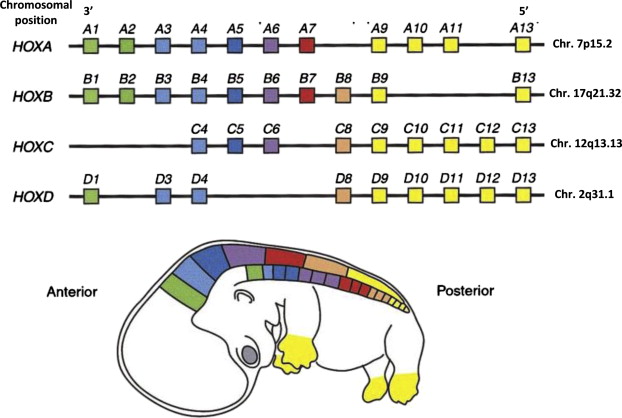
Human HOX gene clusters - Veraksa, A.; Del Campo, M.; McGinnis, W. Developmental Patterning Genes and their Conserved Functions: From Model Organisms to Humans. Mol. Genet. Metab. 2000, 69 (2), 85–100.
Let’s say we want to extract out all the HOX genes from our gene statistics. We can write a regular expression that matches the pattern described above:
dplyr::filter(de_genes, str_detect(hgnc_symbol,"^HOX[A-D][0-9]+$")) %>%
dplyr::arrange(hgnc_symbol)## # A tibble: 39 x 4
## hgnc_symbol mean p padj
## <chr> <int> <dbl> <dbl>
## 1 HOXA1 8734 0.000858 0.00934
## 2 HOXA10 4149 0.00152 0.0102
## 3 HOXA11 567 0.0101 0.0188
## 4 HOXA13 411 0.0105 0.0191
## 5 HOXA2 554 0.00600 0.0151
## 6 HOXA3 18 0.0919 0.0959
## 7 HOXA4 475 0.0113 0.0199
## 8 HOXA5 434 0.0127 0.0211
## 9 HOXA6 3983 0.00252 0.0115
## 10 HOXA7 897 0.00961 0.0183
## # ... with 29 more rowsIn this query we used two new regular expression features:
- within the
[]we specified a range of charactersA-Dand0-9which will match any of the characters between A and D (i.e. A, B, C, or D) and 0 and 9 respectively - the
+character means “match one or more of the preceding expression,” which in our case is the[0-9]. This allows us to match genes with only a single number (e.g.HOXA1) as well as double digit numbers (e.g.HOXA10).
Since we know the cluster identifier part of the HOX gene names (i.e. the
[A-D] part) is exactly one character long, we could alternatively write the
regular expression as follows, using the special . character:
dplyr::filter(de_genes, str_detect(hgnc_symbol,"^HOX.[0-9]+$")) %>%
dplyr::arrange(hgnc_symbol)## # A tibble: 39 x 4
## hgnc_symbol mean p padj
## <chr> <int> <dbl> <dbl>
## 1 HOXA1 8734 0.000858 0.00934
## 2 HOXA10 4149 0.00152 0.0102
## 3 HOXA11 567 0.0101 0.0188
## 4 HOXA13 411 0.0105 0.0191
## 5 HOXA2 554 0.00600 0.0151
## 6 HOXA3 18 0.0919 0.0959
## 7 HOXA4 475 0.0113 0.0199
## 8 HOXA5 434 0.0127 0.0211
## 9 HOXA6 3983 0.00252 0.0115
## 10 HOXA7 897 0.00961 0.0183
## # ... with 29 more rowsHere, the . character is interpreted by the regex to match any single
character, regardless of what it is, between the HOX part and the number
part. This also requires that there exist exactly one character between the two
parts; a gene symbol HOX1 would not be matched, because the 1 would match
to the ., but no number remains to match to the [0-9]+ part.
Sometimes you want to search text for characters that are considered special in the regular expression language. For example, if you had a list of filenames:
filenames <- tribble(
~name,
"annotation.csv",
"file1.txt",
"file2.txt",
"results.csv"
)and wanted to limit to just those with the .txt extension, you need to match
using a literal . character:
filter(filenames, stringr::str_detect(name,"[.]txt$"))## # A tibble: 2 x 1
## name
## <chr>
## 1 file1.txt
## 2 file2.txtInside a [], characters do not have their usual regular expression meaning,
and therefore [.] will match a literal . character. Instead of using the
[] syntax, you may also escape these literal characters using two back
slashes:
filter(filenames, stringr::str_detect(name,"\\.txt$"))## # A tibble: 2 x 1
## name
## <chr>
## 1 file1.txt
## 2 file2.txtRegular expressions are very powerful, and can do much more than what is described here. See the regular expression tutorial linked in the readmore box to learn more details.
5.7.3 dplyr::select() - Subset Columns by Name
Our mutate() operations above created a number of new columns in our tibble,
but we did not specify where in the tibble the new columns should go. Let’s
consider the mutated tibble we created with all four new columns:
dplyr::mutate(gene_stats,
test1_padj=p.adjust(test1_p,method="fdr"),
test2_padj=p.adjust(test2_p,method="fdr"),
signif_either=(test1_padj < 0.05 | test2_padj < 0.05),
signif_both=(test1_padj < 0.05 & test2_padj < 0.05),
gene=stringr::str_to_upper(gene)
)## # A tibble: 3 x 9
## gene test1_stat test1_p test2_stat test2_p test1_padj test2_padj
## <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
## 1 APOE 12.5 0.103 34.2 0.000013 0.155 0.000039
## 2 HOXD1 4.40 0.632 16.3 0.0421 0.632 0.0632
## 3 SNCA 45.7 0.0000000042 0.757 0.915 0.0000000126 0.915
## # ... with 2 more variables: signif_either <lgl>, signif_both <lgl>From a readability standpoint, it might be helpful if all the columns that are about each test were grouped together, rather than having to look at the end of the tibble to find them.
The dplyr::select()
function allows you to pick
specific columns out of a larger tibble in whatever order you choose:
stats <- dplyr::select(gene_stats, test1_stat, test2_stat)
stats## # A tibble: 3 x 2
## test1_stat test2_stat
## <dbl> <dbl>
## 1 12.5 34.2
## 2 4.40 16.3
## 3 45.7 0.757Here we have explicitly selected the statistics columns. dplyr also has helper
functions that allow
for more flexible selection of columns. For example, if all of the columns we
wished to select ended with _stat, we could use the ends_with() helper
function:
stats <- dplyr::select(gene_stats, ends_with("_stat"))
stats## # A tibble: 3 x 2
## test1_stat test2_stat
## <dbl> <dbl>
## 1 12.5 34.2
## 2 4.40 16.3
## 3 45.7 0.757If you so desire, select() allows for the renaming of selected columns:
stats <- dplyr::select(gene_stats,
t=test1_stat,
chisq=test2_stat
)
stats## # A tibble: 3 x 2
## t chisq
## <dbl> <dbl>
## 1 12.5 34.2
## 2 4.40 16.3
## 3 45.7 0.757If we knew that these test statistics actually corresponded to some kind of t-test and a \(\chi\)-squared test, naming the columns of the tibble appropriately may help others (and possibly you) understand your code better.
We can use the dplyr::select() function to obtain our desired column order:
dplyr::mutate(gene_stats,
test1_padj=p.adjust(test1_p,method="fdr"),
test2_padj=p.adjust(test2_p,method="fdr"),
signif_either=(test1_padj < 0.05 | test2_padj < 0.05),
signif_both=(test1_padj < 0.05 & test2_padj < 0.05),
gene=stringr::str_to_upper(gene)
) %>%
dplyr::select(
gene,
test1_stat, test1_p, test1_padj,
test2_stat, test2_p, test2_padj,
signif_either,
signif_both
)## # A tibble: 3 x 9
## gene test1_stat test1_p test1_padj test2_stat test2_p test2_padj
## <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
## 1 APOE 12.5 0.103 0.155 34.2 0.000013 0.000039
## 2 HOXD1 4.40 0.632 0.632 16.3 0.0421 0.0632
## 3 SNCA 45.7 0.0000000042 0.0000000126 0.757 0.915 0.915
## # ... with 2 more variables: signif_either <lgl>, signif_both <lgl>Now the order of our columns is clear and convenient. It is not necessary to list the columns for each test statistic on the same line, but the author thinks this makes the code easier to read and understand.
5.7.4 dplyr::filter() - Pick rows out of a data set
Often, the first step in interpreting an analysis is to identify the features that are significant at some adjusted p-value threshold. First we will save our mutated tibble to another variable, to aid in demonstration:
gene_stats_mutated <- dplyr::mutate(gene_stats,
test1_padj=p.adjust(test1_p,method="fdr"),
test2_padj=p.adjust(test2_p,method="fdr"),
signif_either=(test1_padj < 0.05 | test2_padj < 0.05),
signif_both=(test1_padj < 0.05 & test2_padj < 0.05),
gene=stringr::str_to_upper(gene)
) %>%
dplyr::select(
gene,
test1_stat, test1_p, test1_padj,
test2_stat, test2_p, test2_padj,
signif_either,
signif_both
)Now we can use the dplyr::filter() function to select rows based on whether
they are significant in either test this with our above example.
dplyr::filter(gene_stats_mutated, test1_padj < 0.05)## # A tibble: 1 x 9
## gene test1_stat test1_p test1_padj test2_stat test2_p test2_padj
## <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
## 1 SNCA 45.7 0.0000000042 0.0000000126 0.757 0.915 0.915
## # ... with 2 more variables: signif_either <lgl>, signif_both <lgl>dplyr::filter(gene_stats_mutated, test2_padj < 0.05)## # A tibble: 1 x 9
## gene test1_stat test1_p test1_padj test2_stat test2_p test2_padj
## <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
## 1 APOE 12.5 0.103 0.155 34.2 0.000013 0.000039
## # ... with 2 more variables: signif_either <lgl>, signif_both <lgl>Here we are filtering the result so that only genes with nominal p-value less than 0.05 remain. Note we filter on the two tests separately, but we can also combine these tests using logical operators to achieve different results:
# | means "logical or", meaning the row is retained if either condition is true
dplyr::filter(gene_stats_mutated, test1_padj < 0.05 | test2_padj < 0.05)## # A tibble: 2 x 9
## gene test1_stat test1_p test1_padj test2_stat test2_p test2_padj
## <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
## 1 APOE 12.5 0.103 0.155 34.2 0.000013 0.000039
## 2 SNCA 45.7 0.0000000042 0.0000000126 0.757 0.915 0.915
## # ... with 2 more variables: signif_either <lgl>, signif_both <lgl>Only APOE and SCNA are significant in at least one of the tests.
# & means "logical and", meaning the row is retained only if both conditions are true
dplyr::filter(gene_stats_mutated, test1_padj < 0.05 & test2_padj < 0.05)## # A tibble: 0 x 9
## # ... with 9 variables: gene <chr>, test1_stat <dbl>, test1_p <dbl>,
## # test1_padj <dbl>, test2_stat <dbl>, test2_p <dbl>, test2_padj <dbl>,
## # signif_either <lgl>, signif_both <lgl>It looks like we don’t have any genes that are significant by both tests. Filtering results like this is one of the most common operations we do on the results of biological analyses.
5.7.5 dplyr::arrange() - Order rows based on their values
Another common operation when working with biological analysis results is
ordering them by some meaningful value. Like above, p-values are often used to
prioritize results by simply sorting them in ascending order. The arrange()
function is how to perform this sorting in tidyverse:
stats_sorted_by_test1_p <- dplyr::arrange(gene_stats, test1_p)
stats_sorted_by_test1_p## # A tibble: 3 x 5
## gene test1_stat test1_p test2_stat test2_p
## <chr> <dbl> <dbl> <dbl> <dbl>
## 1 snca 45.7 0.0000000042 0.757 0.915
## 2 apoe 12.5 0.103 34.2 0.000013
## 3 hoxd1 4.40 0.632 16.3 0.0421Note we are sorting by nominal p-value here, not adjusted p-value. In general, sorting by nominal or adjusted p-value results in the same order of results. The only exception is when, due to the way the FDR procedure works, some adjusted p-values will be identical, making the relative order of those tests with the same FDR meaningless. In contrast, it is very rare that nominal p-values will be identical, and since they induce the same ordering of results, when sorting analysis results there are advantages to using nominal p-value, rather than adjusted p-value.
In general, the larger the magnitude of the statistic, the smaller the p-value (for two-tailed tests), so if we so desired we could induce a similar ranking by arranging the data by the statistic in descending order:
# desc() is a helper function that causes the results to be sorted in descending
# order for the given column
dplyr::arrange(gene_stats, desc(abs(test1_stat)))## # A tibble: 3 x 5
## gene test1_stat test1_p test2_stat test2_p
## <chr> <dbl> <dbl> <dbl> <dbl>
## 1 snca 45.7 0.0000000042 0.757 0.915
## 2 apoe 12.5 0.103 34.2 0.000013
## 3 hoxd1 4.40 0.632 16.3 0.0421Here we first apply the base R abs() function to compute the absolute value of
the test 1 statistic and then specify that we want to sort largest first. Note
although we don’t have any negative values in our dataset, we should not assume
that in general, so it is safer for us to be complete and add the absolute value
call in case later we decide to copy and paste this code into another analysis.
That’s pretty much all there is to arrange().
5.7.6 Putting it all together
In the previous sections, we performed the following operations:
- Created new columns by computing false discovery rate on the nominal p-values
using the
dplyr::mutate()andp.adjustfunctions - Created new columns that indicate the patterns of significance for each gene
using
dplyr::mutate() - Mutated the gene symbol case using
stringr::str_to_upperanddplyr::mutate() - Reordered the columns to group related variables with
select() - Filtered genes based on whether they have an adjusted p-value less than 0.05
for either and both statistical tests using
dplyr::filter() - Sorted the results by p-value using
dplyr::arrange()
For the sake of illustration, these steps were presented separately, but
together they represent a single unit of data processing and thus might
profitably be done in the same R command using %>%:
gene_stats <- dplyr::mutate(gene_stats,
test1_padj=p.adjust(test1_p,method="fdr"),
test2_padj=p.adjust(test2_p,method="fdr"),
signif_either=(test1_padj < 0.05 | test2_padj < 0.05),
signif_both=(test1_padj < 0.05 & test2_padj < 0.05),
gene=stringr::str_to_upper(gene)
) %>%
dplyr::select(
gene,
test1_stat, test1_p, test1_padj,
test2_stat, test2_p, test2_padj,
signif_either,
signif_both
) %>%
dplyr::filter(
test1_padj < 0.05 | test2_padj < 0.05
) %>%
dplyr::arrange(
test1_p
)
gene_stats## # A tibble: 2 x 9
## gene test1_stat test1_p test1_padj test2_stat test2_p test2_padj
## <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
## 1 SNCA 45.7 0.0000000042 0.0000000126 0.757 0.915 0.915
## 2 APOE 12.5 0.103 0.155 34.2 0.000013 0.000039
## # ... with 2 more variables: signif_either <lgl>, signif_both <lgl>This complete pipeline now contains all of our manipulations and our mutated tibble can be passed on to downstream analysis or collaborators.