Final Project
For the final project you will develop an R Shiny application that features multiple bioinformatics processes implemented in R. Your application must contain three mandatory components followed by a fourth component of your choice, as described below. You may organize your application however you wish, but it must be integrated into one singular application; four separate applications will not be acceptable. However, it may be helpful to develop each component as its own app, and then combine when all are complete. We suggest a layout like the following:
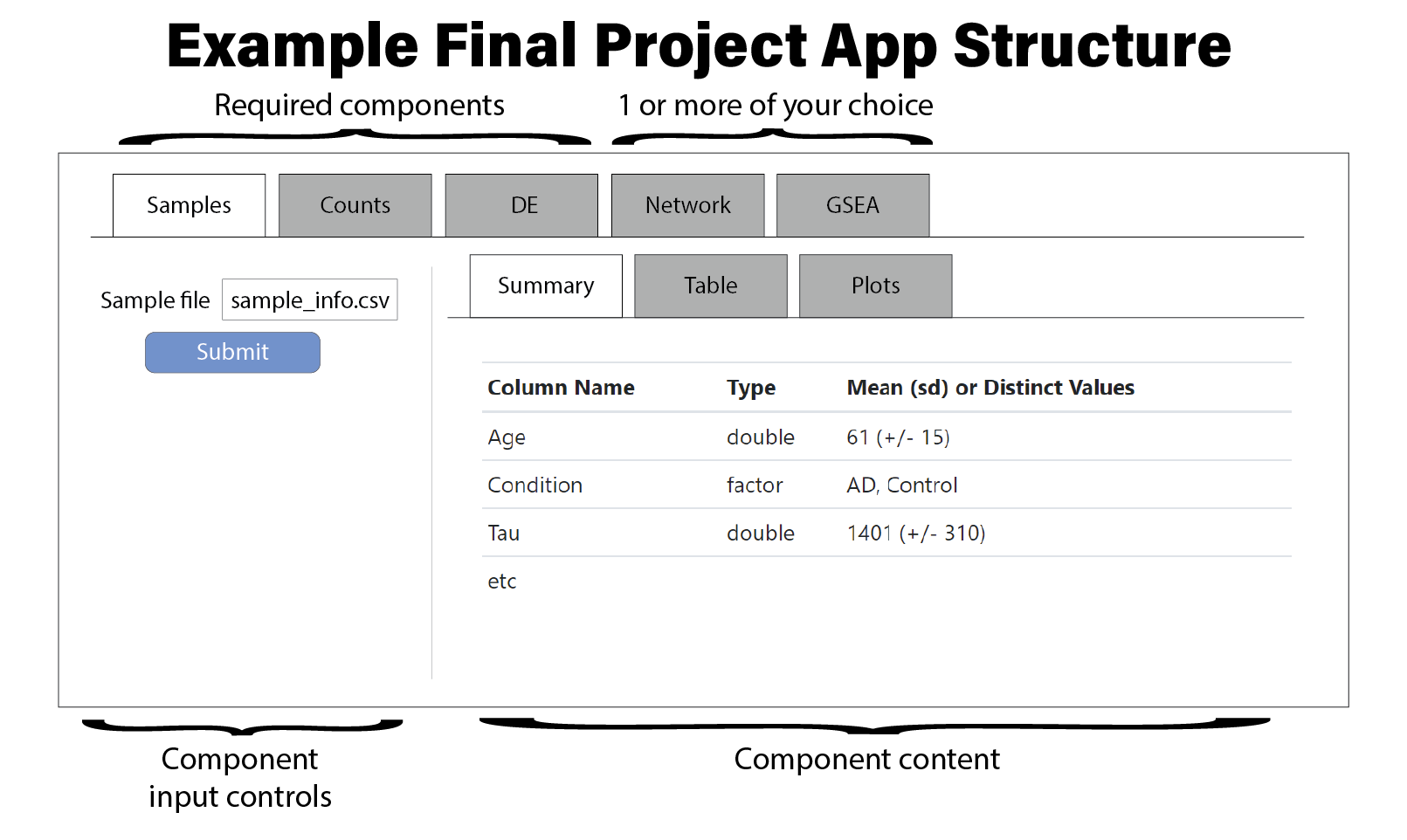
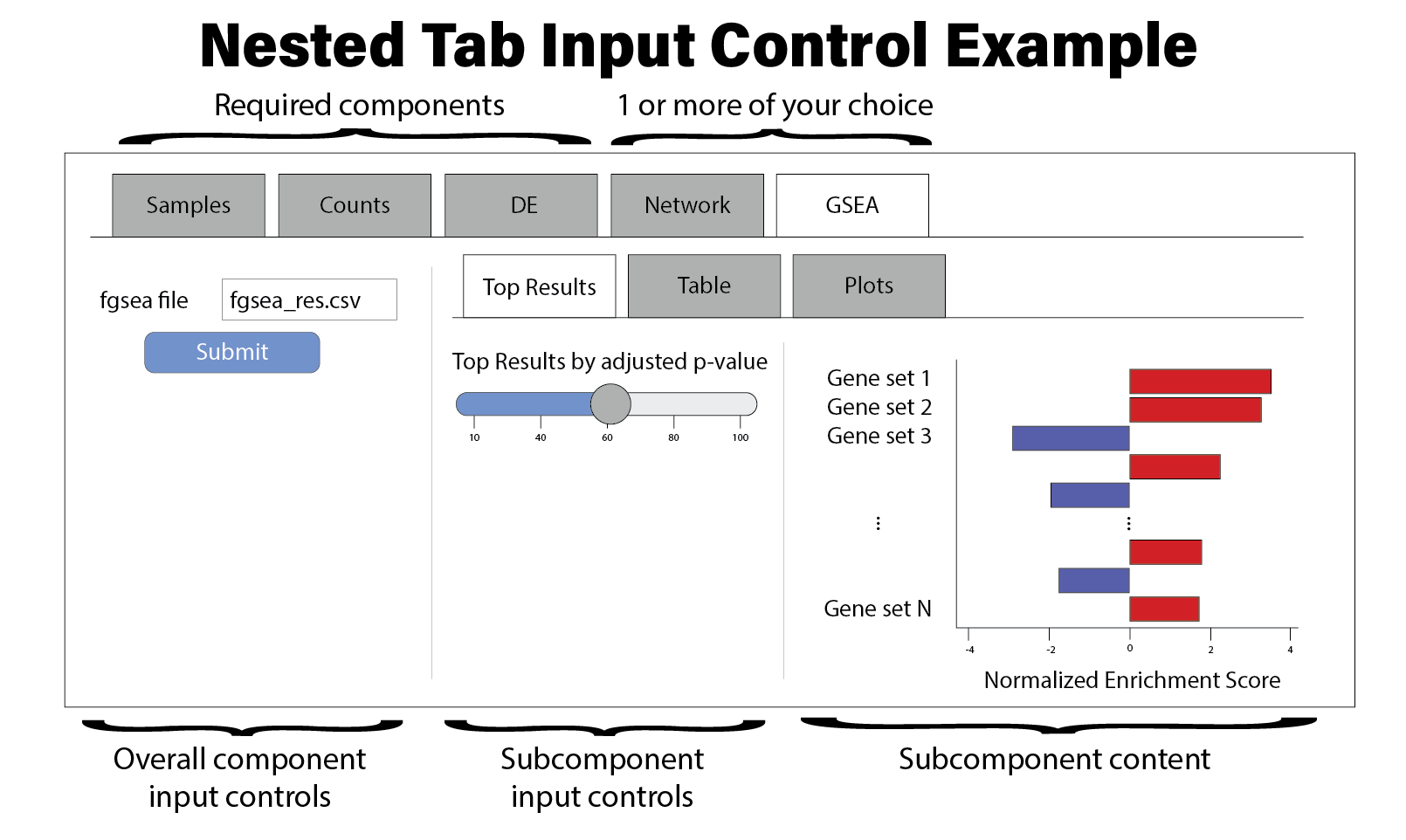
Note some components have subcomponents with their own input controls. You might consider organizing those components as follows:
Part of this project is to identify the data necessary to be loaded into this application. We will provide sets of starting information, but you will need to use the R code you’ve written for assignments one through seven to process the raw data into an appropriate format to be loaded into your Shiny application. Different processing steps and analyses will be required for each dataset, so you must investigate each dataset closely to understand what is available and what must be done yourself.
You must submit your own individual app. This is not a group project. However, you are encouraged to share ideas, troubleshoot and brainstorm together but sharing code is forbidden. Your final submission will include a GitHub repository containing the app and data as well as a five minute pre-recorded screen grab video presentation offering a demonstration of your application. We will view this presentation and grade your project based on how well it fits the following criteria and the contents and quality of your coding.
13.2 General Guidelines
- Simple file input validation (check if it’s CSV or TSV that is well formatted, error message otherwise)
- Write comments in your code elaborating on functions, Shiny commands, or code that is not immediately obvious to someone who isn’t familiar with your code
- Your App should be user friendly. Buttons need to have labels on them. Text descriptions are put in the UI to describe what each section and input is doing.
- (Optional) Data sharing across different tabs. E.g. After analysis is done in the differential expression tab, proceed to the GSEA tab, which will automatically load the results from the differential expression tab.
13.3 Video Presentation Guidelines
- A screen recorded presentation detailing the ins and outs of the Shiny application (Zoom can be used to record)
- Narrate all steps of your presentation as you go
- Presentations should be no more than 5 minutes and should include:
- A brief introduction, tour, and demonstration of each tab in your application
- Discuss some of the challenges and solutions you used while programming
- A very brief explanation of the input data and how it was generated. For example: “The data was comparing control and lung cancer samples from Paper et al. I filtered the counts matrix to only contain genes where no more than half of the samples within each condition had 0 counts. I then performed DE on the counts matrix comparing A vs. B using DESeq2 with default parameters and considered genes to be significant with a FDR < .15”
- Quick walkthrough of Shiny functionalities checklist
We recommend writing a script first to use during your presentation. The average number of spoken words per minute in English is ~140, so you might use that as a guide for length.
13.4 Input Data Sets
We have identified several different datasets that you may use for your projects. Each is a gene expression dataset generated with RNASeq data, and has both sample information and a counts matrix available. You will need to identify the data files containing this information with the references provided below and perform any necessary pre-processing (e.g. counts normalization, differential expression, etc.) to make the data suitable for your app. Each dataset has a slightly different way of obtaining the data, so this should be your first step. The app components described later on this page indicate what the input should be using the language provided here.
Some hints:
- You will need to investigate the data available in each of the provided sources to find sample info. For some of the datasets, it might be easier to simply create a sample info matrix based on filenames, but others will need more detailed (e.g. covariate) information that you will need to find and use.
- You will need to closely examine the available counts matrices to understand what they are and how they have been processed.
- Some of the datasets have precomputed differential expression statistics. You may use these analyses, but be sure you understand what comparison you are using when describing your dataset and demonstrating your app.
You may choose from the following datasets when building and demonstrating your app:
1. Soybean cotyledon gene expression across development
This experiment assayed gene expression in the cotyledons, or embryonic leaves, for soybean plants at different ages after planting.
2. Post-mortem Huntington’s Disease prefrontal cortex compared with neurologically healthy controls
This dataset profiled gene expression with RNASeq in post-mortem human dorsolateral prefrontal cortex from patients who died from Huntington’s Disease and age- and sex-matched neurologically healthy controls.
3. Transcriptional Reversion of Cardiac Myocyte Fate During Mammalian Cardiac Regeneration
This dataset profiled gene expression with RNASeq in murine cardiac tissue across different stages of development to identify genes associated with the loss of these cell’s capacity to regenerate.
4. Molecular mechanisms underlying plasticity in a thermally varying environment
This dataset profiled gene expression with RNASeq in drosophila melanogaster at different stages of development exposed to constant or fluctuating temperatures.
- Molecular mechanisms underlying plasticity in a thermally varying environment
- Salachan, Paul Vinu, and Jesper Givskov Sørensen. 2022. “Molecular Mechanisms Underlying Plasticity in a Thermally Varying Environment.” Molecular Ecology, April. https://doi.org/10.1111/mec.16463.
Note: This article is not open; you will need to find the article through the library of your academic institution
5. Your own idea
You may identify a gene expression dataset of your own interest. The dataset must have:
- A publication associated with it
- A publicly available raw or normalized gene counts matrix with at least 6 samples and 5,000 genes
- Available sample information that allows you to compute a differential expression analysis
Be sure to explain the background of the dataset in your demonstration video.
13.5 Required Components
13.5.1 Sample Information Exploration
The distinct values and distributions of sample information are important to understand before conducting analysis of corresponding sample data. This component allows the user to load and examine a sample information matrix.
Inputs:
- Sample information matrix in CSV format
Shiny Functionalities:
-
Tab with a summary of the table that includes a summary of the type and values in each column, e.g.: Number of rows: X Number of columns: Y
Column Name Type Mean (sd) or Distinct Values Age double 61 (+/- 15) Condition factor AD, Control Tau double 1401 (+/- 310) etc Tab with a data table displaying the sample information, with sortable columns
-
Tab with histograms, density plots, or violin plots of continuous variables.
- If you want to make it fancy, allow the user to choose which column to plot and another column to group by!
13.5.2 Counts Matrix Exploration
Exploring and visualizing counts matrices can aid in selecting gene count filtering strategies and understanding counts data structure. This component allows the user to choose different gene filtering thresholds and assess their effects using diagnostic plots of the counts matrix.
Inputs:
- Normalized counts matrix, by some method or other, in CSV format
- Input controls that filter out genes based on their statistical properties:
- Slider to include genes with at least X percentile of variance
- Slider to include genes with at least X samples that are non-zero
Shiny Functionalities:
- Tab with text or a table summarizing the effect of the filtering, including:
- number of samples
- total number of genes
- number and % of genes passing current filter
- number and % of genes not passing current filter
- Tab with diagnostic scatter plots, where genes passing filters are marked in
a darker color, and genes filtered out are lighter:
- median count vs variance (consider log scale for plot)
- median count vs number of zeros
- Tab with a clustered heatmap of counts remaining after filtering
- consider enabling log-transforming counts for visualization
- be sure to include a color bar in the legend
- Tab with a scatter plot of principal component
analysis projections. You may either:
- allow the user to select which principal components to plot in a scatter plot (e.g. PC1 vs PC2)
- allow the user to plot the top \(N\) principal components as a beeswarm plot
- be sure to include the % variance explained by each component in the plot labels
13.5.3 Differential Expression
Differential expression identifies which genes, if any, are implicated in a specific biological comparison. This component allows the user to load and explore a differential expression dataset.
Inputs:
- Results of a differential expression analysis in CSV format.
- If results are already made available, you may use those
- Otherwise perform a differential expression analysis using DESeq2, limma, or edgeR from the provided counts file
Shiny Functionalities:
- Tab with sortable table displaying differential expression results
- Optional: enable gene name search functionality to filter rows of table
- Tab with content similar to that described in [Assignment 6 / 7]
13.6 Choose-your-own Adventure
Implement one or more of the following components as part of your app.
13.6.1 Gene Set Enrichment Analysis
Use your differential gene expression results to compute gene set enrichment analysis with fgsea. You will need to identify an appropriate gene set database that matches the organism studied in your dataset.
Input:
- a table of fgsea results from the differential expression data.
- choose an appropriate ranking metric (log fold change, -log(pvalue), etc) from your differential expression results
- run fgsea with appropriate parameters against gene set database of your choice
- save the results to a CSV/TSV
- file upload button
Shiny Functionalities:
- Tab 1
- Sidebar
- Slider to adjust number of top pathways to plot by adjusted p-value
- Main Panel
- Barplot of fgsea NES for top pathways selected by slider
- Click on barplot for pathway to display table entry
- Sidebar
- Tab 2
- Sidebar
- Slider to filter table by adjusted p-value (Reactive)
- Radio buttons to select all, positive or negative NES pathways
- Download button to export current filtered and displayed table results
- Main panel
- Sortable data table displaying the results
- Sidebar
- Tab 3
- Sidebar
- Slider to filter table by adjusted p-value (Reactive)
- Main panel
- Scatter plot of NES on x-axis and -log10 adjusted p-value on y-axis, with gene sets below threshold in grey color
- Sidebar
13.6.2 Correlation Network Analysis
Create an app that computes pairwise gene expression correlation for a specific set of input genes. The genes could be all genes in a particular pathway, associated with disease, or perhaps the top \(N\) differentially expressed genes. Subset the gene expression matrix to include only the genes specified in the input text area and compute all pairwise correlations. Your app should report if any of the input genes are not found in the matrix. Also provide a slider input that allows the user to set a minimum threshold for an edge between two genes to be included (e.g. only draw edges for genes with a correlation > 0.9). The slider should be reactive, so the tables and plots update appropriately. Use the [igraph] package as described in the Network Analysis section to construct your graph and to compute network statistics on each node.
Input:
- Normalized counts matrix, by some method or other, in CSV format
- A multi-line text box (hint: textAreaInput) that accepts one gene name per line
- A slider setting the minimum correlation for drawing an edge
Shiny Functionalities:
- Tab with a clustered heatmap of normalized counts corresponding to the genes entered in the text area
- Tab with a visualization of the correlation network
- Vertices should be colored NOT by the default color in igraph
- Vertices should be labeled with gene names
- (Optional) The user can select two vertices, click a button, and show the shortest path
between them. What required in this question are:
- Two drop down menus (selectizeInput) to select two vertices
- Show the shortest path as text output
- Tab with a table with the following metrics calculated for each gene in the input:
- Degree
- Closeness centrality
- Betweenness centrality
13.6.3 RShiny Test Suite
Instead of creating a fourth tab as a part of this project, you will instead create a test application file for use with the “testthat” or “shinytest” packages.
testthat can be used to create a headless (no visual elements) shiny application where you can set inputs and use testthat functions like expect_equal() to compare outputs. shinytest involves manually recording the state of the application and comparing subsequent runs to this state.
Create a test file that uses either or both packages to test various parts of your application. Each tab must have at least 5 distinct tests that measure inputs and outputs of your application (so at least 15 total).
Tests should be relevant and should test parts of the application relevant to
the app’s running. For instance, a test confirming a counts file was filtered
correctly would be acceptable. Tests can measure the output of functions inside
your application or the output$ of the app itself.
Spend a portion of your presentation video running the test to show what is working (or what doesn’t) and discuss briefly your approach to writing and running the tests. Your test file will be reviewed with your project submission.
13.6.4 Visualization of Individual Gene Expression(s)
Visualizing individual gene counts is sometimes useful for examining or verifying patterns identified by differential expression analysis. There are many different ways of visualizing counts for a single gene. This app allows counts from an arbitrary gene to be selected and visualized broken out by a desired sample information variable.
Input:
- Normalized counts matrix, by some method or other, in CSV format
- Sample information matrix in CSV format
- Input control that allows the user to choose one of the categorical fields found in the sample information matrix file
- Input control that allows the user to choose one of the genes found in the counts matrix (hint: try implementing a search box)
- Input control allowing the user to select one of bar plot, boxplot, violin plot, or beeswarm plot
- A button that makes the thing go
Shiny Functionalities:
- Content displaying a plot of the selected type with the normalized gene counts for the selected gene split out by the categorical variable chosen
13.6.5 Custom Option
We have explored a number of different bioinformatics topics in this class and BF528, but there are many more. If you would like to create your own fourth component, draft a small proposal of what you would like your Shiny app to do and what data it will use and send that to Adam or any of the TAs.
General requirements: * Have 3 to 5 input elements (e.g. upload handler, radio selection input, text input, drop down menu, slidebar input, color selection…) * Have 2 to 3 output elements (e.g. plots, table, text…) * Basic preprocessing of the data before input to Shiny app is required. You don’t need to do a comprehensive analysis, but it should not be “download something then use it directly”. You may refer to other sections above to see the amount of pre-processing required. * You may also adopt one of the pre-defined optional tab above (Gene Set Enrichment Analysis, Network Analysis, RShiny Test Suite) with some slight modifications (e.g. a different types of plot)