Lab 5 - CRISPR-Cas9 Guide Selection
Objectives
-
Understand the order of processes necessary to extract a genomic region by its coordinates from an indexed reference
-
Use the UCSC table browser to extract genomic coordinates for important features
-
Finish the
main.nfworkflow declaration to call the provided processes to bgzip the reference fasta, index the fasta, and extract a region by its coordinates. -
Use biopython to try to identify all possible potential guide RNAs in the PKP1 genomic locus
-
Compare what you are able to find with the results from ChopChop
Overview
We discussed CRISPR-Cas9 guide selection in lecture and now you will see the relatively simply bioinformatics work that would enable you to determine the list of potential guides to target Cas9 to a genomic region of interest.
For this lab, remember that the standard Cas9 enzyme can be directed to cleave a target region in the genome with the following conditions:
- The region is a 20nt sequence
- This sequence is immediately followed by a PAM (NGG)
Although there are other considerations that go into optimizing the selection of guide RNAs, these are the two basic requirements.
There are more than a few guide selection algorithms readily available and behind the scenes they all likely perform a few elementary bioinformatics operations you are already familiar with.
The following is the output from one such tool, ChopChop, and lists all potential guide RNAs to target the PKP1 gene in the human genome.
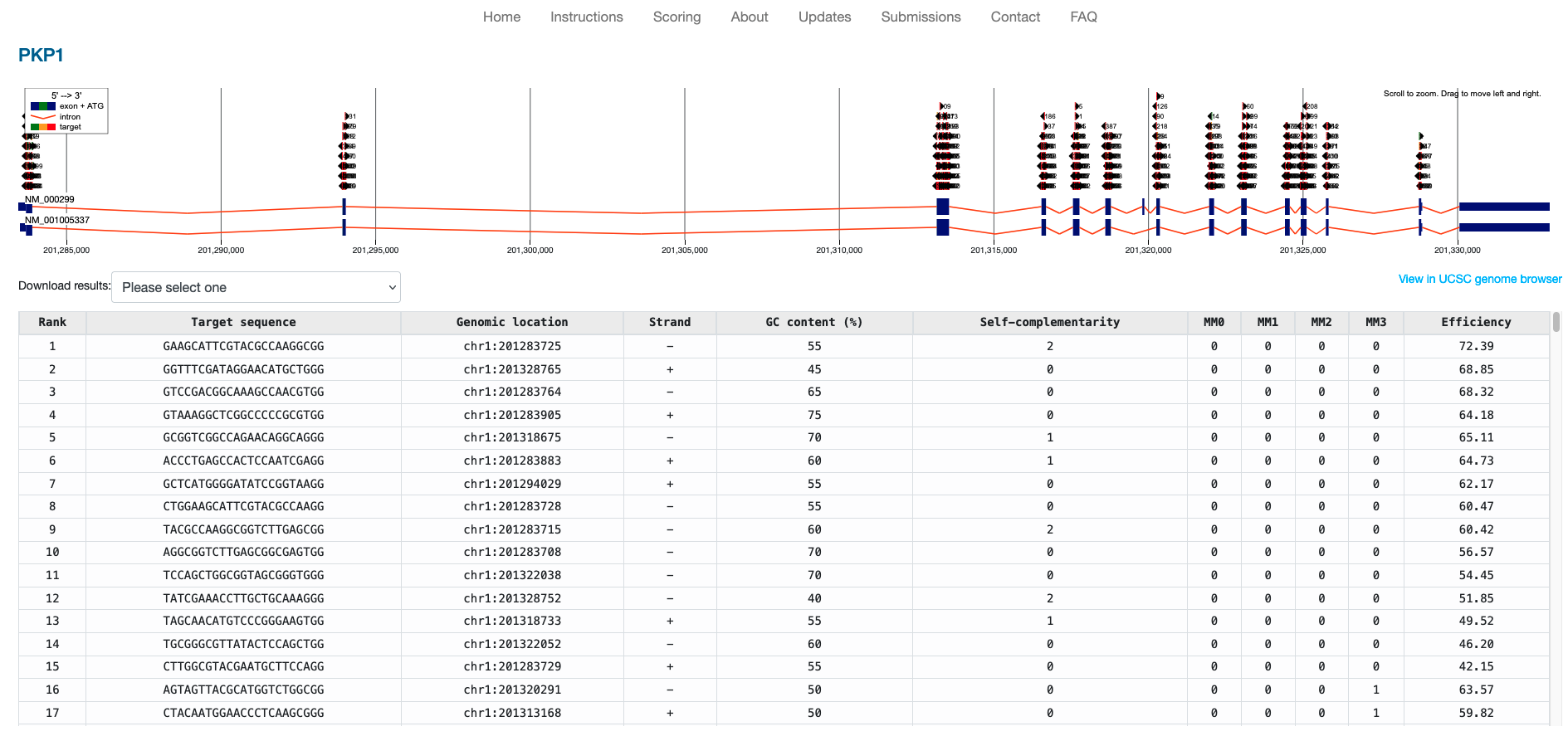
Provided Files
- GRCh38 Gencode Human Reference Genome
- BED file of the start/stop coordinates for all genes in the hg38 reference
- TSV and screenshot of results from ChopChop
- Nextflow modules
Nextflow Activity (15 minutes)
Before you start, please copy the human reference genome and BED file we downloaded
Monday to the refs/ directory you cloned. If you weren’t there, you can find these
files in our class materials (/projectnb/bf528/materials/lab04_template/)
The goal of today is to use the provided modules to create a nextflow workflow that bgzips the reference fasta file, creates an index using samtools, and extracts out a region of interest from the reference.
I have given you all of the code to do so except for the actual workflow. The modules
are fully functional and already have the correct running commands and environments.
Look at the modules you have been provided and particularly take note of the inputs and outputs.
Try to construct a workflow in the main.nf that calls these processes to accomplish
our goal. You may need to adjust some names/paths in the nextflow.config.
Use the following command when you believe you have managed to complete the workflow:
nextflow run main.nf -profile cluster,conda -with-report
BioPython Usage (10 minutes)
We are going to create a conda environment that we will use for our jupyter notebook. This will become a standard practice to also ensure that analyses done in your notebooks are as reproducible and portable as possible.
-
Install the jupyter notebook extension in VSCode if you haven’t already
-
Directly create the conda environment
conda env create -f envs/notebook_env.yml -
Open the jupyter notebook in VSCode and select the environment you just created as its kernel
In your notebook, use Biopython to load in the extracted sequence and attempt to generate code that will identify every possible PAM and valid CRISPR Guide RNA for the extracted sequence.
Similar to ChopChop, please also report the GC content % of each guide as well as its starting position’s genomic coordinates.
I highly encourage you to use any resource available to you, including LLMs, stackoverflow, or each other.
Once you have identified your potential guides, you can compare them to those found in
the results/results.tsv or the screenshot of guides found in this README.
Extra, if you have extra time, see if you can identify the sites on the (-) strand.